 2A+ Farma Portal de notícias
2A+ Farma Portal de notícias


Por Jair Calixto*
Parte 1
Em 21 de agosto de 2019, após muitas audiências públicas e várias reuniões com as empresas e associações do setor industrial farmacêutico, a ANVISA publicou a Resolução RDC nº 301/2019, que trata da revisão das Boas Práticas de Fabricação de medicamentos (BPFs), ancorada pelas normas da PIC/S – Pharmaceutical Inspection Co-operation Scheme (https://www.picscheme.org/)– organismo internacional que publica guias e normas sobre Boas Práticas de Fabricação e Inspeção de medicamentos.
A nova Resolução das BPFs revisou a anterior resolução – RDC nº 17/2010, a qual foi baseada no Technical Report Series nr. 908 da OMS, publicado em 2003.
Esta nova norma atualiza as práticas de fabricação e controle de medicamentos ao nível mundial, estabelecendo similitude com Europa, Japão e Estados Unidos. Isto tem benefícios, mas não é assunto para ser discutido agora.
Neste texto, pretendo mostrar os pontos principais de mudanças trazidas pela resolução e que, de algum modo, irão causar impactos à rotina das empresas.
O capítulo II, Sistema da Qualidade Farmacêutica, é mesmo novo em alguns aspectos, trazendo a definição apresentada no Guia ICH Q 10 – The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) – www.ich.org.
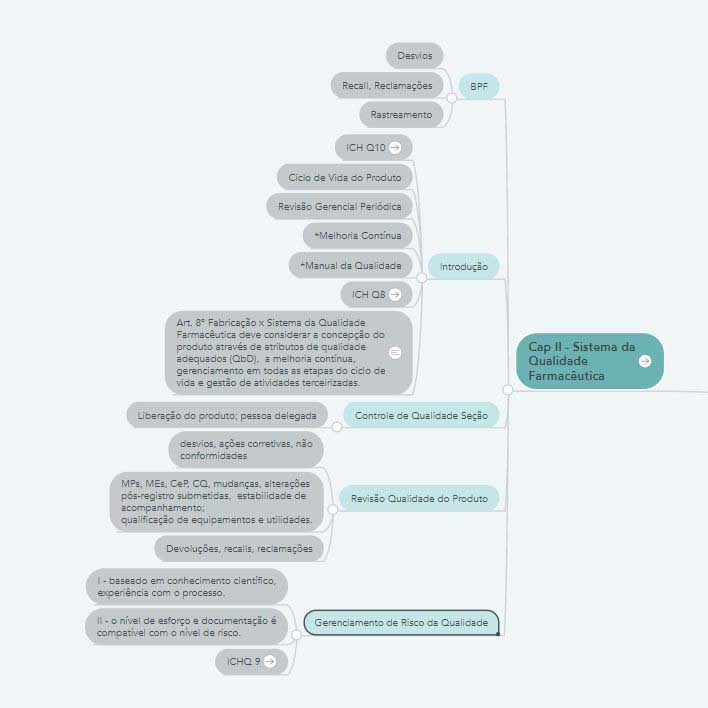
Mapa dos principais novos requisitos de BPF inseridos no Capítulo II da RDC nº 301/2019.
Assim, a ANVISA procurou adequar a forma como as empresas podem gerir as BPFs internamente, conforme a estrutura apresentada no Guia ICH Q10.
Particularmente, considero a seção I – Introdução – uma das mais importantes desta resolução, pois traz temas bem novos para o setor farmacêutico, no Brasil, como Melhoria Contínua, Revisão Gerencial Periódica, Ciclo de Vida do Produto, além de abordar o Manual da Qualidade, já conhecido das empresas. Indiretamente os Guias ICH Q8 e Q10 estão mencionados na resolução.
Os conceitos de Melhoria Contínua terão que ser entendidos, já que não há uma fórmula matemática para isso, nem um roteiro para “fazer” a melhoria. A avaliação crítica das ferramentas existentes dentro dos regulamentos (investigação de desvios, tratamento de reclamações, devoluções, auditorias, recolhimentos, mudanças, revisão periódica do produto, análise de risco) são matérias primas para implantar melhorias contínuas. Contudo, é preciso organizar tudo isso de modo racional.
O Gerenciamento do Ciclo de Vida do Produto vai merecer uma reorganização da gestão da qualidade nas empresas, pois as 4 etapas que o compõem terão que ser integradas dentro de um sistema. Elas são:
- Desenvolvimento farmacêutico.
- Transferência de Tecnologia.
- Fabricação comercial.
- Descontinuação do produto.
Assim, toda a estrutura relacionada a processos, controle de qualidade, formulação, insumos, qualificações, auditorias, validações, desvios de qualidade e gerenciamento dos riscos envolvidos deverão estar dispostos em cada uma das fases e organizados de maneira racional, que possam estar disponíveis em um relatório gerencial para análise, quando necessário.
Entre outros aspectos, o Sistema da Qualidade Farmacêutica, adequado à fabricação de medicamentos, deve garantir:
- que a Concepção do Produto (QbD) por meio de uma estrutura de projeto, planejamento e estabelecimento de um estado de controle, seja consistente a produtos com atributos de qualidade apropriados,
- que o conhecimento de produtos e processos seja gerenciado em todas as etapas do ciclo de vida e
- que se estabeleça uma forma de assegurar a gestão de atividades terceirizadas.
Ainda, nesta seção, foi estabelecida a Revisão Gerencial como forma de envolver a alta gerência com a Melhoria Contínua. A Revisão Gerencial, conforme explicado pela ANVISA, trata-se de uma reunião periódica, com a participação da média e alta gerência com o objetivo de verificar a efetividade do Sistema da Qualidade Farmacêutica da empresa. A revisão deve avaliar os indicadores de desempenho (p.ex. número de reclamações de mercado, número de desvios de qualidade, não conformidades em auditorias) estabelecidos pelo SQF, de modo a proporcionar melhoria constante do sistema, determinando a melhoria da qualidade da empresa como um todo.
Na seção seguinte, sobre as BPFs, destaco a abordagem sobre desvios de qualidade, recolhimentos e reclamações, que por sinal, aparece em outras seções, cabendo uma seção exclusiva para tratar destes pontos – a Seção IX. Portanto, aos desvios de qualidade deve ser dada atenção especial, desde a abertura da investigação, passando pela identificação da causa raiz, mitigação, acompanhamento da ação preventiva e fechamento do relatório.
Na Seção III- Controle de Qualidade – a maioria dos requerimentos estabelecidos já são conhecidos pela área de qualidade das empresas, porém, destaco apenas a ênfase dada para a liberação do produto e a atribuição desta atividade por uma pessoa “Pessoa Delegada pelo Sistema de Gestão da Qualidade”. Note-se que há uma formal responsabilidade atribuída àquela pessoa encarregada de liberar o produto para o mercado. Uma pessoa com autoridade, experiência e entendimento do conjunto de requisitos técnico-regulatórios terá que revisar a documentação de cada lote antes de liberar para a distribuição.
Há uma seção específica sobre Revisão da Qualidade do Produto, muito mais detalhada que na RDC nº 17/2010. A abordagem sobre desvios e ações corretivas são mais profundas e enfáticas. E por que isso? Simplesmente por que os desvios e sua consequente investigação são um dos alicerces de sustentação das melhorias contínuas.
Há, também, um enfoque sobre as mudanças e alterações pós-registro submetidas. Aqui, outro ponto de melhoria contínua, que além do controle, que naturalmente deve ter, também necessita do Gerenciamento de Riscos como ferramenta para validar as mudança e mediar o impacto sobre a qualidade dos medicamentos e a segurança do paciente, à frente.
Outra seção importante, Gerenciamento do Risco na Qualidade – aqui suportado pelo Guia ICH Q9 – é citado com profusão nesta resolução. E não é para menos, já que a ANVISA tem colocado esta ferramenta como sustentáculo para a maioria das novas normas, recaindo para as empresas mais liberdade, porém, com muito mais responsabilidade sobre as decisões em seus processos, produtos e as avaliações rotineiras. Menciono os dois princípios do Gerenciamento de Riscos da Qualidade:
I – a avaliação do risco à qualidade é baseada em conhecimento científico, experiência com o processo e, em última instância, vincula-se à proteção do paciente;
Nota: conhecimento científico e experiência com o processo são fundamentais para realizar análise de risco. Impossível fazer uma análise de risco consistente e robusta sem um mínimo de ciência e domínio sobre o processo. Parece simples, mas não é. Não são muitas as pessoas em uma empresa que dominam completamente o processo farmacêutico, de ponta a ponta. Por esta razão, e, acima de tudo, recomenda-se uma equipe multidisciplinar experiente para a execução da análise de riscos em um processo.
II – o nível de esforço, formalidade e documentação do processo de Gerenciamento de Risco da Qualidade é compatível com o nível de risco.
Nota: quanto maior o potencial de risco, mais detalhes, mais empenho para defini-lo e classificá-lo e mais atenção são necessários para sua Determinação, Controle e Revisão.
Concluindo, portanto, somente no capítulo II da RDC nº 301/2019 foram inseridos diversos novos requisitos para aprimorar a qualidade dos medicamentos dentro do Sistema da Qualidade Farmacêutica. Meu entendimento é que o esforço em caracterizar, entender e implantar cada um destes requisitos tornará o processo farmacêutico mais robusto, mais controlado, com maior conhecimento científico pelas empresas, possibilitando maior flexibilidade na avaliação das situações de rotina.